Published: Dec 23, 2013 by Gavin
If you talk to any chemist, they will tell you the importance of solvent in chemistry. The majority of reactions, especially those in research and used to produce commercial products, take place in the solution phase. It is rare to encounter reactions that are done purely in gas or solid phase. Any article or seminar about organic synthesis will tell you how a reaction works in this solvent but not that one.
Unfortunately, it is quite difficult to model solvent effects using computational chemistry methods. At their most basic, most computational models assume that the system exists at zero kelvin (which is a different problem entirely) in a vacuum. This makes it quite difficult to account for solvent effects. You could simply add solvent molecules to your model system. However, this is generally impractical if you are doing any sort of electronic structure theory calculations, since it can take dozens or hundreds of solvent molecules to achieve complete solvation of a relatively small molecule.
The most basic way to introduce solvent to your calculations is implicit solvent models. There are a number of ways to do this, but the one I’m most familiar with is the self-consistent reaction field (SCRF) method. This method places the model system in a cavity within a dielectric medium and assigns charges to surface of the cavity. It then uses the electric field produced by the charges to polarize the electron density of the model system approximating the electronic effects of solvent. Although this doesn’t completely reproduce solvent effects, it can significantly improve some calculations. The canonical example for this is the alpha amino acids. A standard gas phase calculation predicts that the neutral amino acid is the most stable form. However, when applying an implicit solvent model, the zwitterionic form (with the amine protonated and carboxylic acid deprotonated) is more stable, reproducing experiment. In my own research, applying an implicit solvent model (usually for an aqueous environment) to a reaction will stabilise reactant, product, and transition state structures leading to an overall reduction of the predicted energy barrier by at least 5-10 kJ/mol.
Unfortunately, as alluded to earlier, implicit solvent models are not always sufficient to capture the most important solvent effects in a reaction. In my PhD research, I encountered two instances where inclusion of explicit solvation was required, in one case to dramatically stabilise the transition state and in the other the transition state could not be isolated without the solvent molecules included.
Early on in my PhD program, I was studying the reduction of hydrogen peroxide by organoselenium antioxidants. One of the organoselenium compounds I examined was a diselenide that reacted with the peroxide to form water and Se-O double bond, as shown below.
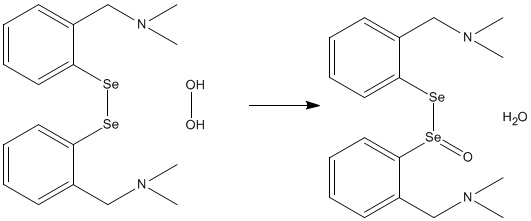
Reaction of the Diselenide Antioxidant with Hydrogen Peroxide
Experimental reaction kinetics suggested that this was a one-step reaction, so I was trying to find a one-step reaction path. I was able to isolate a transition state that showed a concerted reaction with a proton transfer from one oxygen to the other occurring simultaneously with the formation of the Se-O bond. It had a very large energy barrier of 115 kJ/mol, significantly higher than any other antioxidant I modelled in this study. Because of the high barrier, I thought that maybe the transition state was stabilised by hydrogen bonding from the solvent, which experimentally is either water or methanol. I iteratively added a few water molecules to the reaction site and reoptimised the reaction path.
The first water added forms a hydrogen bond the the oxygen that is bonding to the selenium centre. This disappointingly increased the energy barrier by about 8 kJ/mol. With the addition of a second water, the energy barrier dropped to 73 kJ/mol. Examining the structure showed that this water molecule did more than simply hydrogen bond to the peroxide, it acts as a proton shuttle for the hydrogen transfer turning the three-centre transition state into a four-centre one, which is much more stable. A third water molecule added to the model again hydrogen bonds to the other peroxide oxygen, further stabilising the system, giving it an energy barrier of 34 kJ/mol.
A couple of years later, I was now studying the regeneration of the oxidised organoselenium antioxidants, in the form of a selenenic acid (RSe-OH) using thiols (published here). This reaction proceeds via a two-step process, first one thiol reacts with organoselenium molecule, forming a selenylsulfide bond and liberating water. Then a second thiol reacts, forming a disulfide and the reduced selenol (RSeH). In a, perhaps foolish, attempt to avoid requiring the inclusion of solvent, I first modelled this reaction using a deprotonated thiolate to attack the selenenic acid, with a hydroxide liberated rather than water. As one would expect, this had a huge energy barrier of 175 kJ/mol due, at least in part, to the unsuitability of hydroxide as a leaving group.
I relented and attempted to find a transition state for the reaction using the thiol. Trying to isolate a reaction path analogous to the one described above for the diselenide, with simultaneous Se-S bond formation and hydrogen transfer, was unsuccessful. However, after adding two water molecules to the model, a low-energy reaction with identified with both water molecules acting as proton shuttles to facilitate the hydrogen transfer. Following the complete reaction path shows that the three proton transfers are not concerted, but instead follow a “cascade” pattern with the first triggering the second and the second triggering the third, demonstrated in the animation below. This reaction has a barrier of between 40-50 kJ/mol depending on the thiol used.
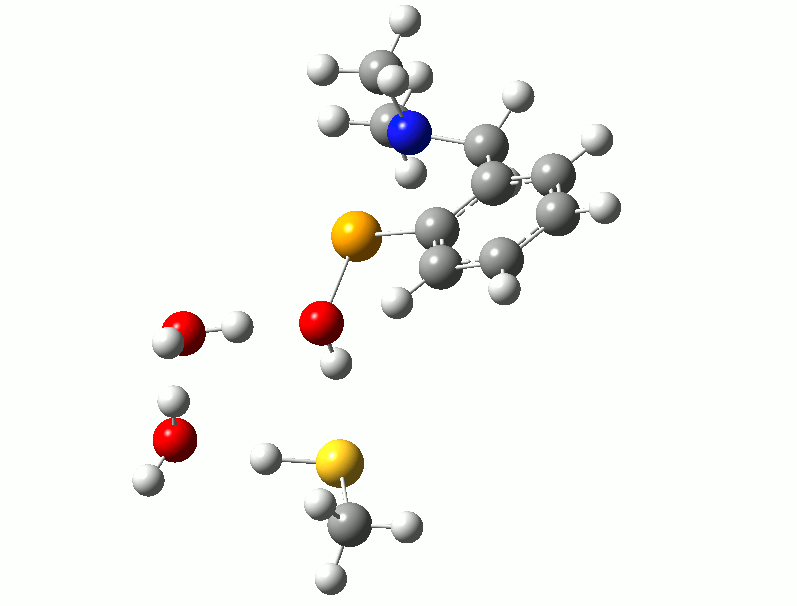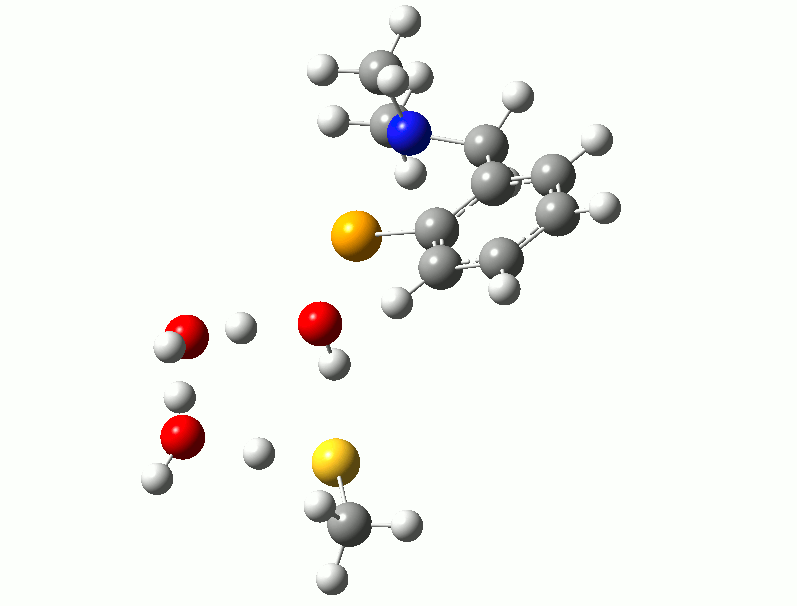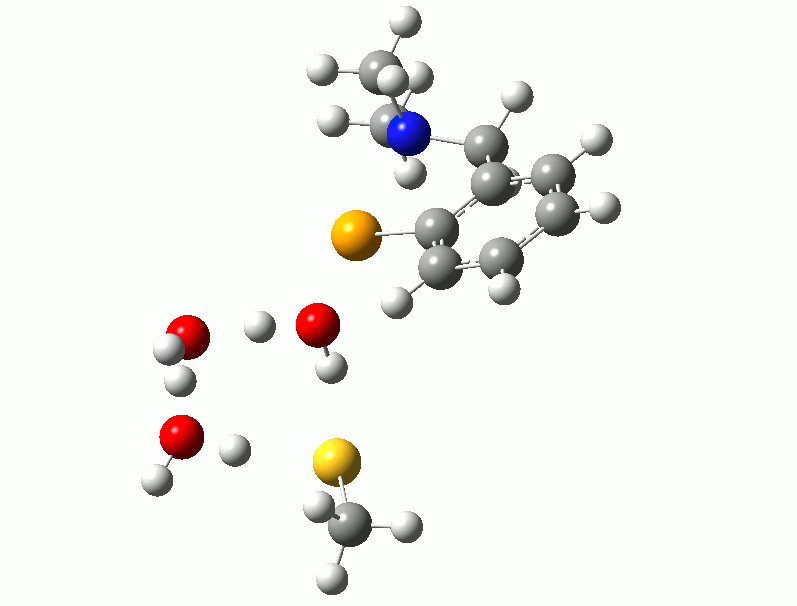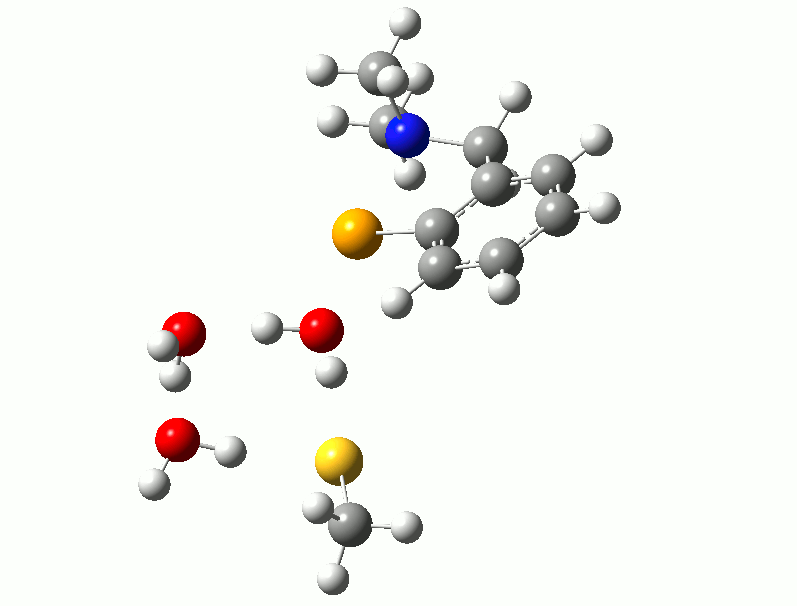
Proton Shuttles - If you don't see animation, try clicking on the image.